












How to Define "Design Review Complete" For Medical Electronics

At a Glance
Learn when a medical device design review is truly complete. Meet FDA standards, ensure traceability, and avoid costly compliance risks.







Design reviews play a critical role in the development and regulatory approval of medical devices. Within the framework of the FDA’s Quality System Regulation (QSR), specifically under 21 CFR Part 820.30, design reviews are not just procedural formalities, they are essential checkpoints that help ensure a device is safe, effective, and meets both user needs and regulatory expectations. These reviews are structured opportunities for cross-functional teams to assess the progress and integrity of a design at various stages of development.
"Design Review Complete" often causes confusion due to varying interpretations across organizations, device complexity, and design process maturity. This ambiguity can lead to inconsistencies, missed requirements, or FDA inspection findings. Thus, a clear, consistent definition is crucial for internal alignment and FDA compliance. This article will clarify the concept using best practices, regulatory guidance, and implementation strategies for medical device manufacturers to ensure thorough and compliant design reviews.
Regulatory Expectations
The FDA has set forth specific expectations regarding design reviews in 21 CFR 820.30(e), which outlines the procedural and documentation requirements that manufacturers must follow. According to this regulation, each design review must be conducted at appropriate stages of the design and development process. These reviews are not optional. They are mandatory checkpoints that help ensure the design is progressing in a controlled and compliant manner.
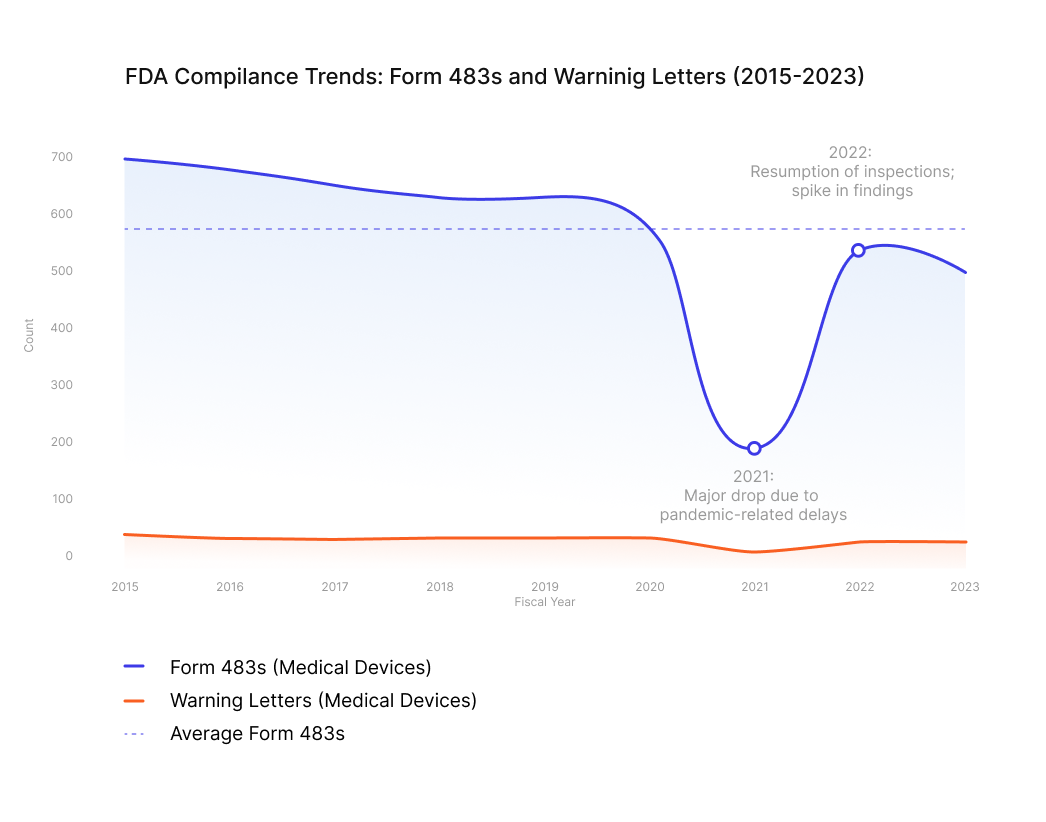
It is estimated that, on average, 570 Form 483’s are issued annually to medical device manufacturers in the United States. This figure underscores the importance of understanding and correctly implementing design review processes in medical electronics. A clear definition of when a design review is considered 'complete' can help engineering and sourcing teams avoid costly delays, rework, and regulatory setbacks.
A key requirement is that the review team must include representatives from all functions that are directly involved in the design stage being evaluated. This typically includes engineering, quality assurance, regulatory affairs, manufacturing, and sometimes marketing or clinical affairs. Importantly, the regulation also mandates the inclusion of at least one individual who does not have direct responsibility for the design stage under review. This person provides an independent perspective, helping to identify potential blind spots or overlooked risks.
The review itself must be comprehensive, meaning it should evaluate the design against established requirements, including user needs, intended use, risk management inputs, and applicable regulatory standards. Any issues identified during the review must be documented, and actions to resolve them should be tracked to closure. The FDA expects that these reviews are not only conducted but also well-documented, with clear records showing who participated, what was discussed, what decisions were made, and how any concerns were addressed.
Failure to meet these expectations can result in Form 483 observations or warning letters, particularly if the FDA determines that design reviews were superficial, poorly documented, or missing altogether. Therefore, it is crucial for organizations to treat design reviews as formal, structured events that contribute directly to the quality and safety of the final product.
Key Indicators of Completion
Determining whether a design review is truly complete requires more than just checking a box or collecting signatures. It involves a thorough evaluation of several critical indicators that reflect the maturity, quality, and compliance of the design process. Below are six key indicators that, when fully addressed, signal that a design review has been completed in accordance with FDA expectations and industry best practices.
1. All design inputs and outputs have been reviewed and documented.
A foundational element of any design review is the confirmation that all design inputs, such as user needs, regulatory requirements, and performance specifications, have been clearly defined, reviewed, and approved. Equally important is the verification that design outputs, including drawings, specifications, and manufacturing instructions, align with those inputs. This alignment must be documented in a traceable manner, often using a requirements traceability matrix (RTM). A complete review ensures that every input has a corresponding output and that any discrepancies have been addressed and resolved.
2. Verification and validation activities are complete, and discrepancies resolved.
Verification and validation (V&V) are critical to demonstrating that the device design meets both its specifications and its intended use. Verification confirms that the design outputs meet the design inputs, typically through inspections, tests, or analyses. Validation, on the other hand, ensures that the final product performs as intended in the actual or simulated use environment. A design review is not complete unless all planned V&V activities have been executed, results have been reviewed, and any non-conformances or test failures have been investigated and resolved. Documentation should include test protocols, results, and evidence of issue resolution.
3. Risk management activities, including FMEA, are finalized.
Risk management is a continuous process throughout the design lifecycle, and its status is a key indicator of review completeness. Tools such as Failure Modes and Effects Analysis (FMEA), Fault Tree Analysis (FTA), and Hazard Analysis should be finalized and reviewed during the design review. This includes ensuring that all identified risks have been evaluated, mitigated to acceptable levels, and documented in accordance with ISO 14971. The review should confirm that risk controls are implemented and verified, and that residual risks are clearly communicated and accepted.
4. Action items from previous reviews are closed.
Design reviews often generate action items, such as requests for additional testing, design changes, or clarifications. A review cannot be considered complete unless all outstanding action items from prior reviews have been addressed, resolved, and documented. This includes verifying that corrective actions were implemented effectively and that any follow-up reviews or approvals have been completed. Maintaining a design review log or action item tracker can help ensure accountability and traceability.
5. The Design History File (DHF) is updated and complete.
The DHF is the official record of the design process and must be kept current throughout the project. It should include all design plans, inputs, outputs, reviews, verification and validation results, risk management documentation, and change records. A complete DHF demonstrates that the design was developed in accordance with the approved procedures and regulatory requirements. During the final design review, the team should confirm that the DHF is comprehensive, organized, and audit ready.
6. Cross-functional sign-off has been obtained.
A design review is not complete without formal approval from all relevant stakeholders. This includes representatives from engineering, quality assurance, regulatory affairs, manufacturing, and any other functions involved in the design process. Additionally, at least one independent reviewer, someone not directly responsible for the design stage, must participate, as required by 21 CFR 820.30(e). Sign-off should be documented using a design review form or checklist, which captures the names, roles, and approval dates of all participants. This ensures that the review reflects a consensus and that all perspectives have been considered.

Practical Guidance for Teams
Successfully completing a design review requires more than just technical expertise. It demands structured planning, cross-functional coordination, and disciplined execution. Engineering, sourcing, and quality teams can significantly improve their readiness for design reviews by adopting a proactive and systematic approach. Below are several practical strategies that teams can implement to ensure design reviews are thorough, timely, and compliant with FDA expectations.
1. Establish clear criteria for each design phase.
Each phase of the design and development process, such as concept, feasibility, design input, design output, verification, and validation, should have well-defined entry and exit criteria. These criteria serve as benchmarks for progress and help teams determine when a phase is ready for review. For example, the design input phase might require finalized user needs, risk assessments, and regulatory requirements before moving forward. By clearly articulating these criteria in the design plan, teams can reduce ambiguity and ensure that reviews are conducted at the right time with the right information.
2. Use checklists aligned with FDA expectations.
Checklists are powerful tools for ensuring consistency and completeness in design reviews. Teams should develop and use standardized review checklists that reflect the requirements of 21 CFR Part 820.30 and incorporate internal quality system procedures. These checklists should cover key areas such as design input/output alignment, risk management, verification and validation, documentation status, and cross-functional participation. Using checklists not only helps reviewers stay focused but also provides a documented record of what was evaluated and approved.
3. Maintain traceability between requirements, design outputs, and test results.
Traceability is a cornerstone of design control and a frequent focus during FDA inspections. Teams should maintain a requirements traceability matrix (RTM) that links user needs and regulatory requirements to specific design outputs and associated test cases. This matrix should be updated throughout the project and reviewed during each design review. Strong traceability ensures that all requirements are addressed, facilitates impact assessments for design changes, and supports verification and validation efforts.
4. Conducti internal audits before formal reviews.
Before initiating a formal design review, teams can benefit from conducting internal audits or peer reviews to identify and resolve issues early. These audits can be informal walkthroughs or structured assessments using the same checklists and criteria that will be used in the formal review. By identifying gaps in documentation, unresolved risks, or missing test data ahead of time, teams can enter the formal review process with greater confidence and fewer delays.
5. Leverag digital tools to manage documentation and collaboration.
Modern product development often involves geographically dispersed teams and complex documentation requirements. Using digital tools such as electronic quality management systems (eQMS), document control platforms, and collaborative project management software can streamline the design review process. These tools help ensure that documents are version-controlled, accessible, and audit-ready. They also facilitate real-time collaboration, task tracking, and automated reminders, reducing the risk of missed steps or miscommunication.

Common Pitfalls and How to Avoid Them
Even well-intentioned teams can encounter obstacles that delay or derail the design review process. Recognizing these common pitfalls, and taking steps to avoid them, can significantly improve review efficiency and regulatory compliance. Below are some of the most frequent issues and practical strategies for addressing them.
Incomplete Documentation
One of the most common reasons for delayed or failed design reviews is missing or incomplete documentation. This may include absent test reports, outdated risk assessments, or unsigned review forms. To avoid this, teams should implement a document readiness checklist and assign clear ownership for each required deliverable. Regular document audits and version control practices can also help ensure that all materials are complete and current before the review begins.
Unresolved Risk Items
Risk management is an ongoing process, and unresolved risks can be a red flag during design reviews. These may include open failure modes in FMEAs, unverified risk controls, or incomplete hazard analyses. Teams should ensure that all risk-related activities are finalized and documented before the review. This includes confirming that residual risks are acceptable, mitigation measures are implemented, and any open issues are clearly justified or resolved.
Lack of Cross-Functional Participation
Design reviews are intended to be collaborative, cross-functional events. When key stakeholders, such as manufacturing, quality, or regulatory representatives, are absent, important perspectives may be missed, leading to gaps in the review. To prevent this, teams should schedule reviews well in advance, communicate expectations clearly, and ensure that all required functions are represented. Including an independent reviewer, as required by the FDA, is also essential for objectivity and compliance.
Poor Traceability
Without clear traceability between design inputs, outputs, and test results, it becomes difficult to demonstrate that the design meets its intended requirements. This can lead to confusion, rework, or even regulatory findings. Teams should maintain and regularly update a traceability matrix and ensure it is reviewed as part of each design review. This matrix should clearly show how each requirement is addressed, tested, and verified.
The most effective way to avoid these issues is to adopt robust project management practices and involve quality assurance (QA) early and consistently throughout the design process. QA can provide guidance on documentation standards, review readiness, and regulatory expectations. Regular team check-ins, milestone reviews, and risk-based planning can also help keep the project on track and ensure that design reviews are completed efficiently and effectively.
Conclusion
Defining "Design Review Complete" for medical electronics is crucial for safety, effectiveness, and regulatory compliance (21 CFR Part 820.30). Clear indicators like finalized documentation, resolved risks, verified traceability, and cross-functional sign-off, alongside proactive planning, standardized tools, and early QA involvement, enhance design control and reduce regulatory risks.
Avoiding pitfalls like incomplete documentation or lack of stakeholder engagement is crucial to prevent delays and ensure compliance. Robust project management and digital collaboration streamline reviews and boost product quality. Ultimately, a well-executed design review is a strategic opportunity to align teams, validate designs, and ensure the product meets user and regulatory expectations. Treating it as a disciplined, collaborative, and value-adding process strengthens quality systems, bringing safer, more effective medical devices to market.

About Author
Related Resources
Related Technical Documentation
Table of Contents
- Regulatory Expectations
- Key Indicators of Completion
- 1. All design inputs and outputs have been reviewed and documented.
- 2. Verification and validation activities are complete, and discrepancies resolved.
- 3. Risk management activities, including FMEA, are finalized.
- 4. Action items from previous reviews are closed.
- 5. The Design History File (DHF) is updated and complete.
- 6. Cross-functional sign-off has been obtained.
- Practical Guidance for Teams
- 1. Establish clear criteria for each design phase.
- 2. Use checklists aligned with FDA expectations.
- 3. Maintain traceability between requirements, design outputs, and test results.
- 4. Conducti internal audits before formal reviews.
- 5. Leverag digital tools to manage documentation and collaboration.
- Common Pitfalls and How to Avoid Them
- Incomplete Documentation
- Unresolved Risk Items
- Lack of Cross-Functional Participation
- Poor Traceability
- Conclusion
Design to Release, Without the Friction
- Keep reviews tied to the right version
- Reduce handoff confusion and rework
- Spot sourcing and release risk earlier
- Work solo, share when needed
Get Started
Thank you, you are now subscribed to updates.



