












The Role of Design Reviews in Regulatory Compliance for Medical Devices

At a Glance
Design reviews are key to FDA and ISO compliance. Reduce risk, improve traceability, and align teams to deliver safe, effective medical devices.







Design reviews are a cornerstone of the FDA’s Quality System Regulation (QSR), specifically outlined in 21 CFR Part 820.30. These reviews are structured checkpoints that play a vital role in ensuring the safety, effectiveness, and regulatory compliance of medical devices. For companies developing medical electronics, where hardware, software, and firmware must work seamlessly together, design reviews are not just a regulatory requirement. They are strategic opportunities to assess progress, identify risks early, and align development efforts with both business goals and regulatory expectations.
Understanding the Regulatory Framework
The FDA mandates that design reviews be conducted at appropriate stages throughout the design and development lifecycle. According to 21 CFR 820.30(e), each review must include representatives from all relevant functions involved in the design stage under review. Importantly, it must also include at least one individual who does not have direct responsibility for that stage. This requirement is intended to promote objectivity and ensure that the review process is not biased by those too close to the work.
The regulation emphasizes that design reviews must be planned, conducted, and documented. This documentation must include the date of the review, the names of participants, and the outcomes or conclusions reached during the session. These records become a critical part of the Design History File (DHF), which is subject to FDA inspection. The DHF serves as a comprehensive record of the design process and is used by inspectors to verify that the device was developed in accordance with regulatory requirements.
Failure to conduct proper design reviews or to maintain adequate documentation is a common source of non-compliance. In fact, deficiencies in design review records are frequently cited in Form 483 observations and warning letters issued by the FDA. These enforcement actions can delay product approvals, damage a company’s reputation, and result in costly remediation efforts.
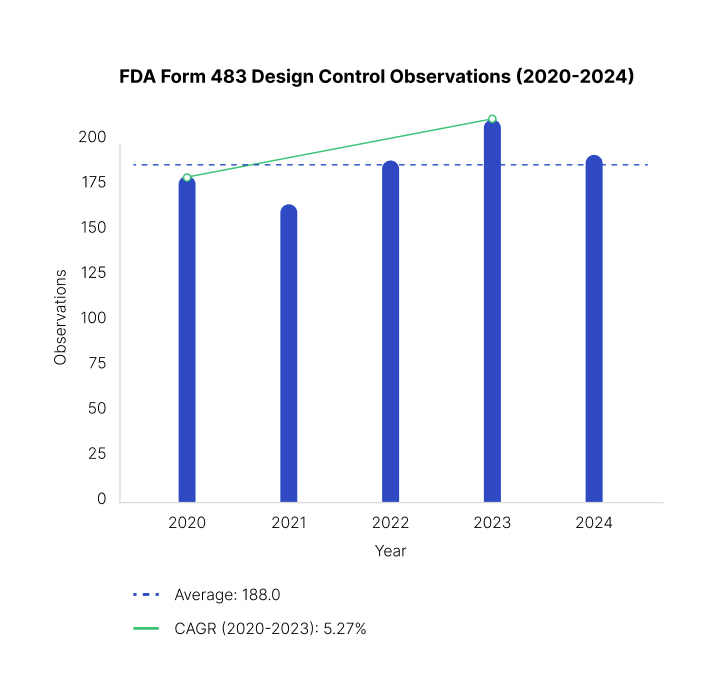
Form 483 observations issued by the FDA related to design control deficiencies have grown by 5% between 2020 and 2023, according to the FDA’s publicly available inspection database. These 21 CFR part 820.30 observations often highlight gaps in design review documentation, traceability, and cross-functional accountability - areas that are critical to regulatory compliance and product safety.
Design reviews also serve as a mechanism for risk management. By evaluating the design at multiple points, teams can identify potential issues before they become costly problems. This proactive approach helps reduce the likelihood of design flaws, manufacturing errors, or post-market failures. It also supports the development of safer, more effective devices that meet both user expectations and regulatory standards.
What Regulators Look For in Design Reviews
Regulatory agencies such as the FDA and international notified bodies closely examine design reviews to ensure they are thorough, well-documented, and inclusive of diverse perspectives. These reviews help confirm a device’s safety, effectiveness, and compliance with applicable standards like 21 CFR Part 820 and ISO 13485:2016 1. Key indicators that regulators look for include:
- Evidence of Risk Management Integration: Regulators expect to see that risk management activities are embedded throughout the design process. This includes updates to tools like Failure Modes and Effects Analysis (FMEA), hazard analyses, and risk control measures. Design reviews should show how risks are identified, assessed, and mitigated, with clear links to design decisions and changes.
- Verification and Validation Planning and Results: Design reviews must include detailed plans for verification and validation (V&V), along with results from completed activities. Verification confirms that design outputs meet design inputs, while validation ensures the final product meets user needs and intended uses. Regulators look for documented test protocols, acceptance criteria, and evidence that any failures were addressed.
- Design Input/Output Traceability: A clear traceability matrix or equivalent documentation should link each design input to corresponding outputs. This ensures that all requirements are met and that changes are tracked. Regulators want to see that the design process is controlled and that nothing critical is overlooked.
- Issue Tracking and Resolution Logs: Design reviews should include logs of identified issues, along with actions taken to resolve them. This includes technical problems, usability concerns, and regulatory gaps. Regulators assess whether issues are tracked systematically, prioritized appropriately, and resolved in a timely manner.
- Independent Reviewer Participation: To ensure objectivity, design reviews must include at least one person who is not solely responsible for the design stage being reviewed. This independent perspective helps uncover blind spots and ensures that the review is not biased by those too close to the work.
Best Practices for Engineering and Sourcing Teams
To meet and exceed regulatory expectations, engineering and sourcing teams should adopt structured, proactive practices that support compliance and product quality. These practices help ensure that design reviews are meaningful, efficient, and audit-ready.
1. Schedule Design Reviews at Logical Phase Gates
Design reviews should be planned at key milestones in the development process, such as concept freeze, prototype completion, and design freeze. These phase gates allow teams to assess progress, make informed decisions, and ensure readiness for the next stage.
2. Use Checklists Aligned with 21 CFR 820.30 and ISO 13485
Standardized checklists help ensure that all required elements are covered during each review. These checklists should reflect both FDA and ISO requirements, including risk management, design controls, and documentation standards. They also help teams stay consistent across projects.
3. Ensure Cross-Functional Participation Including Quality, Regulatory, and Manufacturing
Effective design reviews bring together experts from multiple disciplines. Quality assurance, regulatory affairs, manufacturing, and sometimes clinical or marketing teams should be involved. This diversity ensures that all aspects of the product—technical, regulatory, and operational—are considered.
4. Document Action Items and Follow-Ups Rigorously
Every design review should result in a list of action items, assigned owners, and due dates. Follow-up reviews should confirm that these actions were completed. This documentation shows regulators that the team is responsive and committed to continuous improvement.
5. Maintain Version-Controlled Design Review Records in the DHF
All design review documents should be stored in the Design History File (DHF) with proper version control. This includes meeting minutes, attendance records, review materials, and outcomes. Regulators expect to see a clear audit trail that reflects the evolution of the design, and the decisions made along the way.
Common Pitfalls and How To Avoid Them
Many companies struggle with design reviews because they lack a structured approach or fail to document decisions properly. These gaps can lead to regulatory issues, missed risks, and costly delays. Below are common pitfalls and practical ways to avoid them:
- Treating Design Reviews as Informal Meetings: One of the most frequent mistakes is conducting design reviews like casual team check-ins. Without a formal agenda, clear objectives, and documented outcomes, these meetings lose their value. Regulators expect structured reviews with defined scope, roles, and deliverables. To avoid this, establish a formal review process with scheduled milestones, standardized templates, and clear expectations for each participant.
- Failing to Include Independent Reviewers: Design reviews must include at least one person who is not directly involved in the design stage being reviewed. This independent perspective helps uncover blind spots and ensures objectivity. Skipping this step can lead to biased evaluations and missed issues. To prevent this, assign independent reviewers from other departments or external consultants, and rotate them to maintain fresh insights.
- Inadequate Documentation of Decisions and Rationale: Many teams document what was discussed but not why decisions were made. This lack of rationale makes it hard to justify choices during audits or future reviews. Regulators want to see the reasoning behind design changes, risk mitigations, and technical trade-offs. To improve, use structured templates that prompt teams to record not just decisions, but the supporting data, risk assessments, and stakeholder input that led to those decisions.
- Poor Traceability Between Design Inputs and Outputs: Traceability is essential for demonstrating that all user needs and regulatory requirements have been addressed. When inputs (like user requirements or regulatory standards) are not clearly linked to outputs (like specifications or test results), it creates gaps in compliance. To fix this, implement a traceability matrix that maps each input to its corresponding output, and update it throughout the design process.
Case Study Insights from Regulators
1. FDA Warning Letters and 483 Observations
The FDA regularly publishes Form 483 inspection observations and warning letters that serve as real-world case studies. Many of these cite failures in design control, particularly around design reviews.
- Example: A 2023 FDA warning letter to a U.S. medical device manufacturer cited the company for failing to include independent reviewers in their design reviews and for not documenting the rationale behind design decisions. This led to a Class II recall due to a software malfunction that could have been caught earlier.
2. EMA – EU COMBINE Project (2024)
The EU COMBINE project, led by the European Medicines Agency (EMA), analysed the challenges of aligning clinical trials, performance studies, and medical device investigations under the MDR, IVDR, and CTR frameworks.
- Key Insight: The project found that lack of coordination and structured documentation in design and clinical review processes led to delays in trial approvals and regulatory confusion across member states.
- Relevance: This case reinforces the need for harmonized well-documented design reviews that integrate risk management and usability from the start.
3. EMA Guideline on Drug-Device Combinations (2022)
This EMA guideline outlines quality documentation expectations when a medicinal product is used with a medical device. It includes examples of non-compliance due to poor traceability and inadequate design review documentation.
- Key Insight: Several submissions were delayed or rejected because companies failed to demonstrate how design inputs (e.g., user needs, regulatory requirements) were translated into design outputs and verified through structured reviews.
Conclusion
Design reviews are not just regulatory requirements. They are essential tools for ensuring the safety, effectiveness, and compliance of medical devices. When conducted properly, they provide structured opportunities to evaluate progress, identify risks, and align cross-functional teams. Regulatory bodies like the FDA and EMA expect these reviews to be thorough, well-documented, and inclusive of diverse perspectives. They look for unmistakable evidence of risk management, traceability, independent review, and actionable outcomes. Case studies and inspection findings consistently show that companies who treat design reviews as strategic checkpoints are better positioned to avoid recalls, delays, and compliance issues.
Informal meetings, missing documentation, and lack of traceability jeopardize the design process, leading to regulatory citations, product failures, or costly rework. These pitfalls are avoidable by establishing design review protocols, training teams on regulations, and using standardized tools. Cross-functional participation, version-controlled documentation, and clear action tracking are crucial safeguards for innovation and compliance. Real-world examples from FDA warning letters and EMA reports demonstrate that structured, well-documented design reviews are essential. Learning from these cases and applying proven strategies strengthens processes, reduces risk, and delivers safer, more effective medical devices to market.

About Author
Related Resources
Related Technical Documentation
Table of Contents
- Understanding the Regulatory Framework
- What Regulators Look For in Design Reviews
- Best Practices for Engineering and Sourcing Teams
- 1. Schedule Design Reviews at Logical Phase Gates
- 2. Use Checklists Aligned with 21 CFR 820.30 and ISO 13485
- 3. Ensure Cross-Functional Participation Including Quality, Regulatory, and Manufacturing
- 4. Document Action Items and Follow-Ups Rigorously
- 5. Maintain Version-Controlled Design Review Records in the DHF
- Common Pitfalls and How To Avoid Them
- Case Study Insights from Regulators
- 1. FDA Warning Letters and 483 Observations
- 2. EMA – EU COMBINE Project (2024)
- 3. EMA Guideline on Drug-Device Combinations (2022)
- Conclusion
Design to Release, Without the Friction
- Keep reviews tied to the right version
- Reduce handoff confusion and rework
- Spot sourcing and release risk earlier
- Work solo, share when needed
Get Started
Thank you, you are now subscribed to updates.